Recursive feature elimination coupled to SVM in R
I recently wondered whether it is possible to apply Recursive Feature Selection with SVM in R. RFE+SVM is well described in the literature. It has originally been proposed by Guyon and coworkers for gene selection, in Gene Selection for Cancer Classification using Support Vector Machines (Machine Learning (2002) 46(1)3), 389-422). Here are two more recent papers that show application of RFE+SVM in a similar context and discuss its performance as a wrapper method for feature selection:
- Niijima, S. and Kuhara, S. (2006). Recursive gene selection based on maximum margin criterion: a comparison with SVM-RFE. Bioinformatics, 7: 543.
- Ding, Y. and Wilkins, D. (2006). Improving the Performance of SVM-RFE to Select Genes in Microarray Data. BMC Bioinformatics, 7(Supp. 2): S12.
I discovered the pathClass package, that implements standard RFE+SVM and reweighted RFE described in Johannes, M., Brase, J.C. Fröhlich, H., Gade, S., Gehrmann, M., Fälth, M., Sültmann, H., and Beißbarth, T. (2010). Integration of pathway knowledge into a reweighted recursive feature elimination approach for risk stratification of cancer patients. Bioinformatics, 26(17): 2136–2144.
It relies on kernlab, which is pretty good because it supports a larger set of kernel functions and is well maintained. Also, Alex Smola, who co-authored the package, has written with B. Schölkopf an awesome reference textbook1 on Learning with Kernels (MIT Press, 2002, see Chapter 1 for an overview).
From the vignette, here is an example of use on a 72 x 7129 dataset where the outcome is AML (n=25) or ALL (n=47) in the Golub study [1].
library(pathClass)
library(golubEsets)
data(Golub_Merge)
y <- pData(Golub_Merge)$ALL
x <- exprs(Golub_Merge)
x <- t(x)
res.rfe <- crossval(x, y, DEBUG=TRUE, theta.fit=fit.rfe, folds=10,
repeats=5, parallel=TRUE, Cs=10^(-3:3))
extractFeatures(res.rfe, toFile=FALSE)
plot(res.rfe, toFile=FALSE)
The crossval() function above performs 10-fold cross-validation (repeated 5 times) using RFE for feature selection. Be careful: It is rather long! Also, please note that we didn’t use the graph structure for features connectivity (protein-protein interactions), nor did we take advantage of annotation facilities in R (i.e., for mapping probe set ID to protein ID).
The ROC curve of the AUC for each repeat is shown below (left):
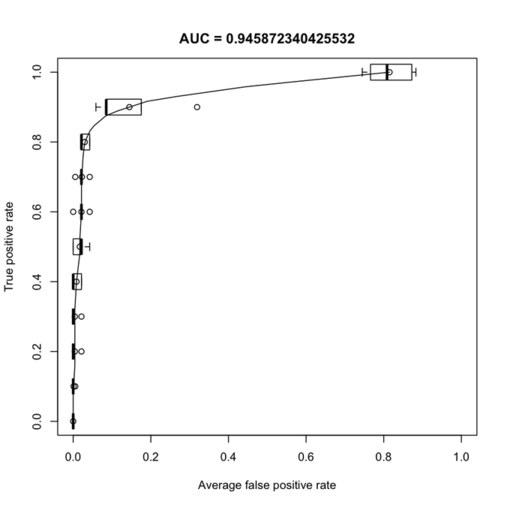
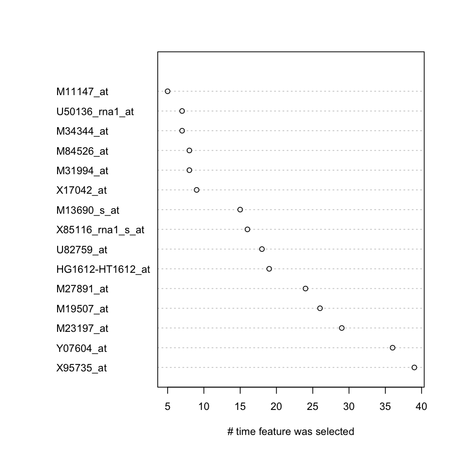
On the right in the figure above, I show the top 15 genes retained by the feature selection procedure.
More information can be found in the vignette, pathClass: SVM-based classification with prior knowledge on feature connectivity.
By the way, I just discovered that HarlanH provided very nice code for displaying classification results.
References
- Golub, T.R., Slonim, D.K., Tamayo, P., Huard, C., Gaasenbeek, M., Mesirov, J.P., Coller, H., Loh, M.L., Downing, J.R., Caligiuri, M.A., Bloomfield, C.D., and Lander, E.S. (1999). Molecular Classification of Cancer: Class Discovery and Class Prediction by Gene Expression Monitoring, Science, 531-537.
Another textbook I like is Bakir, G.H., Hofmann, T., Schölkopf, B., Smola, A.J., Taskar, B., and Vishwanathan, S.V.N. (2006). Predicting Structured Data. MIT Press. ↩︎