Méthodes biostatistiques
ANOVA et comparaisons
multiples
Table of Contents
Les paramètres suivants doivent être définis dans R ou
RStudio afin de reproduire les analyses présentées dans ce
document. Les packages ggplot2,
hrbrthemes, directlabels,
Hmisc et multcomp ne font pas
partie de la distribution R et doivent être installés au
préalable si nécessaire :
install.packages(c("ggplot2", "hrbrthemes", "directlabels", "Hmisc", "multcomp"))
Les dépendances de ces packages seront installées
automatiquement. On supposera également que les
instructions R sont exécutées dans un répertoire de travail
avec les fichiers de données accessibles dans un
sous-répertoire data/. Si ce n’est pas le cas,
il suffit de créer un sous-répertoire data/ et
d’y enregistrer les fichiers de données, ou de redéfinir
les chemins d’accès dans les instructions de lecture des
fichiers de données ci-après.
library(ggplot2) library(hrbrthemes) library(directlabels) library(Hmisc) library(multcomp) options(digits = 6, show.signif.stars = FALSE) theme_set(theme_ipsum(base_size = 11))
1 Rappels sur l’ANOVA à un facteur
Soit \(y_{ij}\) la $j$ème observation dans le groupe \(i\). Le modèle d’ANOVA ou “effect model” s’écrit \[ y_{ij}=\mu+\alpha_i+\varepsilon_{ij}, \] où \(\mu\) désigne la moyenne générale, \(\alpha_i\) l’effet du groupe \(i\), et \(\varepsilon_{ij}\sim \mathcal{N}(0,\sigma^2)\) un terme d’erreur aléatoire. On impose généralement que \(\sum_{i=1}^k\alpha_i=0\).
L’hypothèse nulle se lit \(H_0:\alpha_1=\alpha_2=\dots=\alpha_k\). Sous cette hypothèse d’égalité des moyennes de groupe, la variance entre groupe (“between”) et la variance propre à chaque groupe (“within”) permettent d’estimer \(\sigma^2\). D’où le test F d’égalité de ces deux variances. Sous \(H_0\), le rapport entre les carrés moyens inter et intra-groupe (qui estiment les variances ci-dessus) suit une loi F de Fisher-Snedecor à \(k-1\) et \(N-k\) degrés de liberté.
1.1 Chargement des données
On utilise les données sur le polymorphisme
du gène du récepteur estrogène, polymorphism.dta.
Le chargement des données au format Stata
nécessite le package foreign et
peut être réalisé comme suit :
d <- foreign::read.dta("data/polymorphism.dta")
str(d)
'data.frame': 59 obs. of 3 variables: $ id : num 1 2 3 4 5 6 7 8 9 10 ... $ age : num 43 47 55 57 61 63 63 69 70 72 ... $ genotype: Factor w/ 3 levels "1.6/1.6","1.6/0.7",..: 1 1 1 1 1 1 1 1 1 1 ... - attr(*, "datalabel")= chr "" - attr(*, "time.stamp")= chr "16 Apr 2001 14:58" - attr(*, "formats")= chr [1:3] "%9.0g" "%9.0g" "%9.0g" - attr(*, "types")= int [1:3] 102 102 102 - attr(*, "val.labels")= chr [1:3] "" "" "rflp" - attr(*, "var.labels")= chr [1:3] "ID Number" "Age at Diagnosis" "Genotype" - attr(*, "version")= int 7 - attr(*, "label.table")=List of 1 ..$ rflp: Named int [1:3] 1 2 3 .. ..- attr(*, "names")= chr [1:3] "1.6/1.6" "1.6/0.7" "0.7/0.7"
1.2 Description des variables
Pour résumer la structure de données assez
simple, on peut calculer l’âge moyen selon les
groupes à l’aide de aggregate et
représenter graphiquement les distributions
conditionnelles de l’âge. Cela dit, le package
Hmisc permet de calculer des
résultats plus détaillés et plus facilement
exploitables. On utilise la fonction,
summary.formula, que l’on peut
abréger à summary comme dans le
cas du résumé d’un data frame ou d’une
variable, et les mêmes arguments que dans le
cas de aggregate.
summary(age ~ genotype, data = d, fun = smean.sd)
age N= 59 +--------+-------+--+-------+-------+ | | | N| Mean| SD| +--------+-------+--+-------+-------+ |genotype|1.6/1.6|14|64.6429|11.1811| | |1.6/0.7|29|64.3793|13.2595| | |0.7/0.7|16|50.3750|10.6388| +--------+-------+--+-------+-------+ | Overall| |59|60.6441|13.4943| +--------+-------+--+-------+-------+
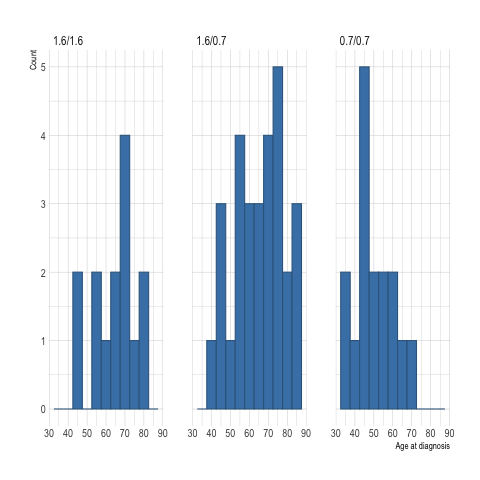
Pour représenter les distributions
conditionnelles, il est possible d’utiliser des
histogrammes d’effectif, des courbes de densité
ou des boîtes à moustaches. Voici une solution
avec des histogrammes à l’aide de
ggplot2 :
p <- ggplot(data = d, aes(x = age)) +
geom_histogram(binwidth = 5, fill = "steelblue", color = "steelblue4") +
facet_wrap(~ genotype, ncol = 3) +
labs(x = "Age at diagnosis", y = "Count")
p
1.3 Modélisation
La commande aov permet de
décomposer la variance totale entre les
différentes sources de variations
(nécessairement de type factor)
indiquées dans le modèle et le terme d’erreur
ou “résiduelle”. On l’utilise généralement en
combinaison avec summary.aov pour
produire le tableau d’ANOVA :
m <- aov(age ~ genotype, data = d)
summary(m)
Df Sum Sq Mean Sq F value Pr(>F)
genotype 2 2316 1158 7.86 0.00098
Residuals 56 8246 147
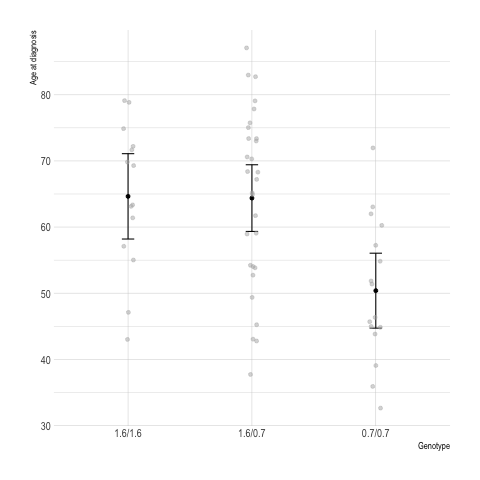
Une représentation graphique sous forme de
moyennes conditionnelles s’obtient assez
facilement avec ggplot2. Le calcul
des moyennes et des intervalles de confiance à
95 % est réalisé avec Hmisc
(attention, summarize n’accepte
pas de notation par formule, contrairement à la
fonction summary.formula du même
package) :
r <- with(d, summarize(age, genotype, smean.cl.normal))
p <- ggplot(data = r, aes(x = genotype, y = age)) +
geom_errorbar(aes(ymin = Lower, ymax = Upper), width=.1) +
geom_point() +
geom_jitter(data = d, width = .05, color = grey(.7), alpha = .5) +
labs(x = "Genotype", y = "Age at diagnosis")
p
On peut comparer l’ensemble des paires de moyennes à l’aide de simples tests de Student non protégés :
with(d, pairwise.t.test(age, genotype, p.adj = "none"))
Pairwise comparisons using t tests with pooled SD
data: age and genotype
1.6/1.6 1.6/0.7
1.6/0.7 0.947 -
0.7/0.7 0.002 5e-04
P value adjustment method: none
Notons que l’option par défaut retenue pour
pairwise.t.test est de travailler
avec la variance commune de l’ensemble des
groupes, comme dans l’ANOVA. Pour retrouver les
mêmes résultats avec la commande
t.test, il faudra désactiver cette
option :
with(d, pairwise.t.test(age, genotype, p.adj = "none", pool.sd = FALSE))
t.test(age ~ genotype, data = d, subset = genotype != "0.7/0.7", var.equal = TRUE)
Pairwise comparisons using t tests with non-pooled SD
data: age and genotype
1.6/1.6 1.6/0.7
1.6/0.7 0.946 -
0.7/0.7 0.001 4e-04
P value adjustment method: none
Two Sample t-test
data: age by genotype
t = 0.06408, df = 41, p-value = 0.949
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
-8.04237 8.56947
sample estimates:
mean in group 1.6/1.6 mean in group 1.6/0.7
64.6429 64.3793
Les mêmes résultats peuvent se retrouver à partir du modèle linéaire directement. Encore une fois, il faut s’assurer que les variables catégorielles sont bien représentées comme des facteurs sous R :
m <- lm(age ~ genotype, data = d)
summary(m)
Call:
lm(formula = age ~ genotype, data = d)
Residuals:
Min 1Q Median 3Q Max
-26.379 -8.643 0.625 8.621 22.621
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 64.643 3.243 19.93 <2e-16
genotype1.6/0.7 -0.264 3.949 -0.07 0.9470
genotype0.7/0.7 -14.268 4.441 -3.21 0.0022
Residual standard error: 12.1 on 56 degrees of freedom
Multiple R-squared: 0.219, Adjusted R-squared: 0.191
F-statistic: 7.86 on 2 and 56 DF, p-value: 0.000978
Le test d’ensemble pour le modèle indiqué au
bas des résultats précédents correspond au test
de Fisher-Snedecor de l’ANOVA. On peut
également le retrouver en affichant le tableau
d’ANOVA du modèle de régression à l’aide de
anova :
anova(m)
Analysis of Variance Table
Response: age
Df Sum Sq Mean Sq F value Pr(>F)
genotype 2 2316 1157.9 7.863 0.000978
Residuals 56 8246 147.2
2 Modèle d’ANOVA à deux facteurs
Soit \(y_{ijk}\) la $k$ème observation pour le niveau \(i\) du facteur \(A\) (\(i=1,\dots,a\)) et le niveau \(j\) du facteur \(B\) (\(j=1,\dots,b\)). Le modèle complet avec interaction s’écrit \[ y_{ijk} = \mu + \alpha_i + \beta_j + \gamma_{ij} + \varepsilon_{ijk},\] où \(\mu\) désigne la moyenne générale, \(\alpha_i\) (\(\beta_j\)) l’écart à la moyenne des moyennes de groupe pour le facteur \(A\) (\(B\)), \(\gamma_{ij}\) les écarts à la moyenne des moyennes pour les traitements \(A\times B\), et \(\varepsilon_{ijk}\sim \mathcal{N}(0,\sigma^2)\) la résiduelle. Les effets \(\alpha_i\) et \(\beta_j\) sont appelés effets principaux, tandis que \(\gamma_{ij}\) est l’effet d’interaction. Les hypothèses nulles associées sont \[ \left\{\begin{array}{lr} H_0^A:\alpha_1=\alpha_2=\dots=\alpha_a, & (a-1)\,\text{dl}\\ H_0^B:\beta_1=\beta_2=\dots=\beta_b, & (b-1)\,\text{dl}\\ H_0^{AB}:\gamma_{11}=\gamma_{13}=\dots=\gamma_{ab}, &(a-1)(b-1)\,\text{dl}\\ \end{array}\right. \]
Des tests F (CM effets / CM résiduelle) permettent de tester ces hypothèses. À noter : des sommes de carré de type I sont estimées pour \(A\), puis \(B\), et enfin \(A\times B\), à la différence des SC de type II/III, mais elles sont égales dans le cas d’un plan complet équilibré.
2.1 Chargement des données
On utilise les données sur le gain de poids chez des rats soumis à différents régimes alimentaires, weight.rda. Le chargement des données est réalisé ci-dessous :
load("data/weight.rda")
str(weight)
'data.frame': 10 obs. of 4 variables: $ V1: int 90 76 90 64 86 51 72 90 95 78 $ V2: int 73 102 118 104 81 107 100 87 117 111 $ V3: int 107 95 97 80 98 74 74 67 89 58 $ V4: int 98 74 56 111 95 88 82 77 86 92
2.2 Description des variables
Les variables peuvent être décrites par colonnes mais il est préférable de réarranger le tableau sous forme de data frame, afin d’avoir une seule variable statistique par colonne :
d <- data.frame(weight = as.numeric(unlist(weight)),
type = gl(2, 20, labels = c("Beef", "Cereal")),
level = gl(2, 10, labels = c("Low", "High")))
head(d)
weight type level 1 90 Beef Low 2 76 Beef Low 3 90 Beef Low 4 64 Beef Low 5 86 Beef Low 6 51 Beef Low
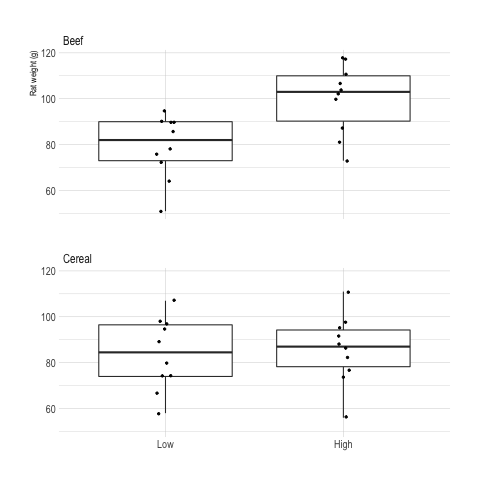
Voici un résumé graphique possible des
données individuelles avec le package
ggplot2 :
p <- ggplot(data = d, aes(x = level, y = weight)) +
geom_boxplot(position = position_dodge()) +
geom_jitter(size = .8, width = .05) +
facet_wrap(~ type, nrow = 2) +
labs(x = NULL, y = "Rat weight (g)")
p
2.3 Diagramme d’interaction
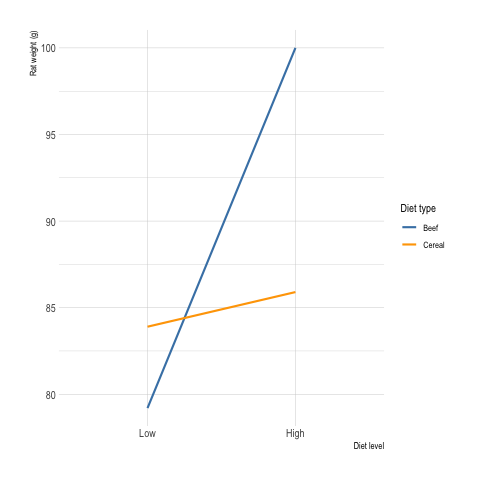
Pour visualiser l’interaction entre les deux variables, on peut utiliser un simple tracé de moyennes conditionnelles :
p <- ggplot(data = d, aes(x = level, y = weight, color = type)) +
stat_summary(fun.y = mean, geom = "line", aes(group = type), size = 1) +
scale_color_manual("Diet type", values = c("steelblue", "orange")) +
labs(x = "Diet level", y = "Rat weight (g)")
p
Une autre manière de procéder consisterait à
travailler avec le data frame de données
agrégées, r, et utiliser
geom_line au lieu de
stat_summary, toujours en veillant
à ajouter une esthétique de groupement. La
fonction geom_errorbar permet de
rajouter aisément des barres d’erreur
(écart-type, erreur-type ou intervalles de
confiance – à calculer séparément), comme on
l’a vu plus haut. Les statistiques nécessaires
peuvent être calculées avec les packages
plyr, dplyr ou
Hmisc comme proposé plus haut.
Voici un exemple avec des intervalles de
confiance à 95 % :
with(d, summarize(weight, llist(type, level), smean.cl.normal))
type level weight Lower Upper
2 Beef Low 79.2 69.2660 89.1340
1 Beef High 100.0 89.1721 110.8279
4 Cereal Low 83.9 72.6626 95.1374
3 Cereal High 85.9 75.1540 96.6460
Le package Hmisc propose aussi
des fonctions graphiques pour représenter,
entre autres, les effets d’interaction.
2.4 Modélisation
Le modèle d’ANOVA à deux facteurs avec interaction s’écrit :
m1 <- aov(weight ~ type * level, data = d)
summary(m1)
Df Sum Sq Mean Sq F value Pr(>F)
type 1 221 221 0.99 0.327
level 1 1300 1300 5.81 0.021
type:level 1 884 884 3.95 0.054
Residuals 36 8049 224
La notation type * level est
équivalente à type + level +
type:level, où type:level
désigne l’interaction entre les facteurs
type et level. Ce
terme d’interaction n’étant pas significatif,
il est possible de proposer un modèle plus
simple :
m0 <- aov(weight ~ type + level, data = d)
summary(m0)
Df Sum Sq Mean Sq F value Pr(>F)
type 1 221 221 0.91 0.345
level 1 1300 1300 5.38 0.026
Residuals 37 8933 241
On retrouvera le test de l’interaction en
comparant ces deux modèles emboîtés,
m0 et m1, à l’aide de
anova (dans le cas gaussien, ce
sont des statistiques F qui sont calculées)
:
anova(m0, m1)
Analysis of Variance Table Model 1: weight ~ type level Model 2: weight ~ type * level Res.Df RSS Df Sum of Sq F Pr(>F) 1 37 8933 2 36 8049 1 883.6 3.952 0.0545
Puisque la variable level
possède deux niveaux, il est également possible
de retrouver à peu près les mêmes résultats que
ceux du tableau d’ANOVA à l’aide d’un test de
Student puisque l’effet de ce facteur est
supposé être indépendant de l’autre terme
inclus dans le modèle :
r <- t.test(weight ~ level, data = d, var.equal = TRUE)
r
r$statistic^2
Two Sample t-test
data: weight by level
t = -2.323, df = 38, p-value = 0.0256
alternative hypothesis: true difference in means is not equal to 0
95 percent confidence interval:
-21.33588 -1.46412
sample estimates:
mean in group Low mean in group High
81.55 92.95
t
5.39495
2.5 Comparaisons multiples
La fonction pairwise.t.test est
limitée au cas à un facteur. On peut néanmoins
reconstruire le terme d’interaction à l’aide de
interaction, ce qui nous fournira
un facteur codant les groupes issus du
croisement de l’ensemble des niveaux des deux
facteurs, encore appelé traitements. Cette
technqiue permet également de réaliser un test
d’égalité des variances ou de visualiser la
distribution entre traitements pour vérifier
les conditions de validité du modèle.
Voici un exemple de tests de Student avec correction de Bonferroni :
with(d, pairwise.t.test(weight, interaction(type, level), p.adj = "bonf"))
Pairwise comparisons using t tests with pooled SD
data: weight and interaction(type, level)
Beef.Low Cereal.Low Beef.High
Cereal.Low 1.00 - -
Beef.High 0.02 0.13 -
Cereal.High 1.00 1.00 0.25
P value adjustment method: bonferroni
Et voici pour la distribution du poids des rats entre traitements :
p <- ggplot(data = d, aes(x = interaction(type, level, sep = "/"), y = weight)) +
geom_boxplot() +
labs(x = "Treatment (Diet type x Diet level)", y = "Rat weight (g)")
p
L’alternative consiste à utiliser des tests
post-hoc de type Tukey HSD à l’aide de
TukeyHSD, ou de l’alternative
disponible via la fonction
multcomp::glht(). Voici une
illustration avec le package
multcomp, plus flexible et plus
générale que la fonction de base de R. À noter
qu’il est également nécessaire de travailler
avec le terme d’interaction et d’utiliser
lm au lieu de aov
:
d$tx <- with(d, interaction(type, level))
m <- lm(weight ~ tx, data = d)
r <- glht(m, linfct = mcp(tx = "Tukey"))
summary(r)
Simultaneous Tests for General Linear Hypotheses
Multiple Comparisons of Means: Tukey Contrasts
Fit: lm(formula = weight ~ tx, data = d)
Linear Hypotheses:
Estimate Std. Error t value Pr(>|t|)
Cereal.Low - Beef.Low == 0 4.70 6.69 0.70 0.895
Beef.High - Beef.Low == 0 20.80 6.69 3.11 0.018
Cereal.High - Beef.Low == 0 6.70 6.69 1.00 0.749
Beef.High - Cereal.Low == 0 16.10 6.69 2.41 0.094
Cereal.High - Cereal.Low == 0 2.00 6.69 0.30 0.991
Cereal.High - Beef.High == 0 -14.10 6.69 -2.11 0.170
(Adjusted p values reported -- single-step method)
Ces résultats vont dans le même sens que ce
que suggère le résultat non significatif pour
le test de l’interaction (cf. les deux
comparaisons extrêmes Cereal.Low -
Beef.Low et Cereal.High -
Beef.High).
3 Exercices d’application
Dans cette application, on travaillera avec le
data frame ToothGrowth qui contient
des données issues d’un plan d’expérience dans
lequel on s’intéresse à la croissance des
odontoblastes de cochons d’inde auxquels on
administre de la vitamine C sous forme d’acide
ascorbique ou de jus d’orange à différentes doses
(0.5, 1 et 2 mg/jour).
Les données sont disponibles dans le package
datasets. Il n’est pas forcément
nécessaire de les charger dans l’espace de travail
mais par souci de clarté on indique la commande
requise :
data(ToothGrowth) summary(ToothGrowth)
Voici une vue d’ensemble des données individuelles et agrégées :
r <- aggregate(len ~ dose + supp, data = ToothGrowth, mean)
p <- ggplot(data = ToothGrowth, aes(x = dose, y = len, color = supp)) +
geom_point(position = position_jitterdodge(jitter.width = .1, dodge.width = 0.25)) +
geom_line(data = r, aes(x = dose, y = len, color = supp)) +
scale_color_manual(values = c("steelblue", "orange")) +
guides(color = FALSE) +
geom_dl(aes(label = supp), method = list("smart.grid", cex = .8)) +
labs(x = "Dose (mg/day)", y = "Length (oc. unit)")
p
3.1 Test de linéarité
Attention au codage de de la variable
dose.
- Réaliser une ANOVA à deux facteurs pour
répondre aux questions suivantes : (a)
existe t-il un effet du mode
d’administration de la vitamine C
(
supp) sur la longueur des odontoblastes, et (b) cet effet est-il dépendant du dosage journalier ? - L’effet dose est-il linéaire ? Vérifier
la linéarité de la relation
len ~ doseen utilisant (a) une approche de régression et (b) une approche par ANOVA avec des contrastes polynomiaux.
3.2 Comparaisons post-hoc
On souhaite comparer l’ensemble des paires
de moyennes pour le croisement des deux
variables supp et
dose.
- Indiquer les résultats obtenus avec une approche par tests de Student sans correction pour les tests multiples.
- Idem mais en contrôlant pour le risque d’expérience global (FWER) par (a) une technique d’ensemble (Bonferroni ou Sidák) et (b) une technique par étape (Holm). Les conclusions diffèrent-elles ?
3.3 Spécification de contrastes
On souhaite comparer des groupes spécifiques d’observations.
- On s’intéresse à la comparaison des
données moyennes dans le groupe
OJpour les doses 0.5 et 1 mg réunies avec les données moyennes dans le groupeVCpour les mêmes doses. Réaliser le test correspondant. - On s’intéresse à la comparaison des
données moyennes dans le groupe
OJpour les doses 0.5 et 1 mg réunies avec les données moyennes dans le groupeOJpour la dose 2 mg. Réaliser le test correspondant. - On s’intéresse à la comparaison des
données moyennes dans le groupe
OJpour les doses 0.5 et 1 mg réunies avec les données moyennes dans le groupeVCpour les doses 1 et 2 mg. Réaliser le test correspondant.
3.4 Type de sommes de carré
R utilise des sommes de carré de type I,
encore appelées séquentielles. Dans le cas où
les effectifs sont balancés entre les
traitements, on obtiendra les mêmes résultats
quelle que soit l’approche utilisée. En cas de
déséquilibre d’effectifs, en revanche, ce n’est
plus le cas et certains auteurs recommendent
d’utiliser des sommes de carré de type III,
voire de type II. Comparer les résultats
obtenus en utilisant ces trois types de sommes
de carré, obtenus à l’aide des fonctions
add11 ou drop1 et
Anova du package car
pour le modèle à deux facteurs sur les données
ToothGrowth. Le déséquilibre
d’effectifs pourra être imposé par une
suppression aléatoire de 10 % des
observations.
set.seed(101) ToothGrowth[sample(1:nrow(ToothGrowth), 6),"len"] <- NA