Phylogenetic analysis using Python
Lately I have been playing with Python and the ETE toolkit to build and compare phylogenetic trees. As discussed in a recent post, phylogenetic trees are used to unravel the evolutionary relationships or lineage between a set of genetic sequences. The ETE toolkit has a long history now,1 and it is definitely the way to go if you are diving into phylogenetic analysis in Python. It is both a module that you can import the usual way in a Python console, and a set of executable workflows that you run from your preferred shell.
We will start by building a database of orthologous genes for 4 species of Mycoplasma. This is easily carried out using Orthofinder, and the example dataset bundled with this package. A detailed tutorial on how to do your own analysis from Ensembl is also available. Assuming you are in the root directory where you installed Orthofinder, just run orthofinder.py and inspect the results directory:
$ python3 orthofinder.py -f ExampleDataset
$ cd ExampleDataset/OrthoFinder/Results_Mar12
$ ls
Citation.txt Log.txt Phylogenetically_Misplaced_Genes WorkingDirectory
Comparative_Genomics_Statistics Orthogroup_Sequences Resolved_Gene_Trees
Gene_Duplication_Events Orthogroups Single_Copy_Orthologue_Sequences
Gene_Trees Orthologues Species_Tree
Note that a little quality check should have been printed near the end of the output. In this case, I got:
Writing orthogroups to file
---------------------------
OrthoFinder assigned 2207 genes (80.8% of total) to 603 orthogroups. Fifty percent of all genes
were in orthogroups with 4 or more genes (G50 was 4) and were contained in the largest 280 orthogroups
(O50 was 280). There were 269 orthogroups with all species present and 246 of these consisted entirely
of single-copy genes.
A tree in Newick format should also be available in the Species_Tree/ subdirectory. Using Python, we can visualize it using the command line utility (ete3), e.g.:
$ ete3 view -t Species_Tree/SpeciesTree_rooted.txt
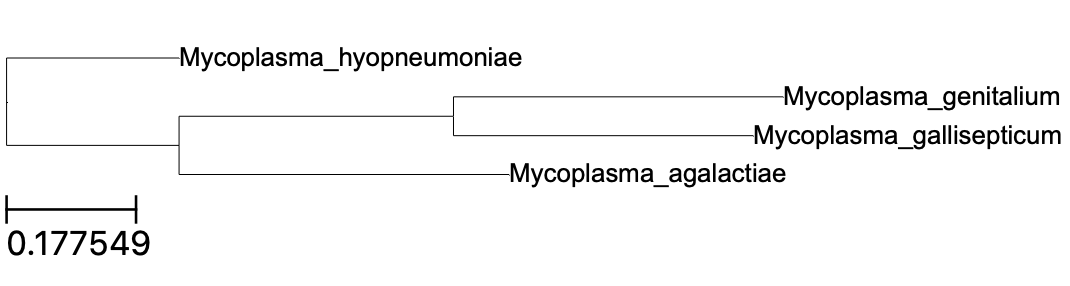
Note, however, that is the “final” species tree, and not gene trees that could potentially be produced on aligned sequences (but see the Gene_Trees subdirectory). Fortunately, all orthologous sequences are available in Fasta format in the Single_Copy_Orthologue_Sequences subdirectory. Two examples of such a gene tree are shown below (click to enlarge).


In principle it is possible to rebuild these trees manually but we need to align the protein sequences first. In the figure above, I considered the orthogroup 7 and 9. In the subdirectory Orthogroup_Sequences/, we can find the corresponding multi-fasta files. Here is a preview of the first aa for the 7th orthogroup:
In [1]: from Bio import SeqIO
In [2]: with open("OG0000007.fa") as f:
...: seqs = SeqIO.parse(f, "fasta")
...: for s in seqs:
...: print(">{}\n{}".format(s.name, s.seq[:20]))
>gi|290753066|emb|CBH41042.1|
MANKDYYKILGVDKKASDKE
>gi|31541124|gb|AAP56426.1|
MRWLVKEQNYYEILGVSTNA
>gi|284811924|gb|AAP56500.2|
MSSKRDYYEILEVSRSATQQ
>gi|284811927|gb|ADB96855.1|
MFQTKKTCPSCKGEGQTIKN
>gi|284811928|gb|ADB96856.1|
MSSKRDYYEILGVSRSATQQ
>gi|31541331|gb|AAP56632.1|
MRPFLESNYYELLGVSETAS
>gi|31541333|gb|AAP56634.1|
MADKQQPTQLNLIAYFDDYQ
>gi|31541383|gb|AAP56684.1|
MIKSEDFMAEKRDYYEVLQI
>gi|3844628|gb|AAC71235.1|
MAAGKRDYYEVLGISKNASS
>gi|3844797|gb|AAC71418.1|
MAEQKRDYYEVLGITPDADQ
>gi|71851550|gb|AAZ44158.1|
MAKQDFYKILGVEKSASLTE
We can perform a multiple alignment of those sequences using MAFFT, and build a tree directly using ETE3 and the Fasttree or RaxML workflow:
$ mafft OG0000007.fa > OG0000007_a.fa
$ gsed -i -e 's/|/-/' OG0000007_a.fa
$ ete3 build -a OG0000007_a.fa -w none-none-none-raxml_default -C 12 -o OG0000007 --clearall
$ ete3 view -t OG0000007/OG0000007_a.fa.final_tree.nw
gsed is macOS equivalent of GNU sed from Homebrew.
In the above statement, none-none-none-raxml_default means that we use no aligner, no trimmer, no (model) tester, and the RaxML utility with default settings. Here’s the output of our manual tree reconstruction:

As can be seen, there’s some room for improvement since the alignment appears quite bad. That being said, we can still compare this tree with the one built by Orthofinder using ete3 compare. We will first need to match the labels in both trees:
$ cd OG0000007
$ cp ../../Gene_Trees/OG0000007_tree.txt .
$ gsed -i 's/|/-/g' OG0000007_tree.txt
$ gsed -i -E 's/Mycoplasma_[a-z]+_//g' OG0000007_tree.txt
$ ete3 compare -t none-none-none-raxml_default/OG0000007_a.fa.final_tree.nw -r OG0000007_tree.txt
Jaime Huerta-Cepas, Joaquín Dopazo and Toni Gabaldón. ETE: a Python Environment for Tree Exploration. BMC Bioinformatics 2010, 11:24. ↩︎